- Mega построение филогенетических деревьев
- Построение и анализ дерева, содержащего паралоги
- Сайт Волынкиной Инны
- Скобочная формула дерева
- Изображение дерева
- Нетривиальные ветви дерева
- Реконструкция филогении
- Phylogenetic Tree Construction Made Easy with Blast & Mega
- Blast
- Multiple Sequence Alignment (MSA)
- Which One to Choose, ClustalW or MUSCLE?
- Phylogenetic Tree Construction with MEGA Version 6
- Tree Explorer
- References:
Mega построение филогенетических деревьев
В задании требовалось построить филогенетическое дерево выбранных бактерий, используя последовательности РНК малой субъединицы рибосомы (16S rRNA). Искомые последовательности были добыты из *.frn файлов, находящихся в базе полных геномов NCBI. Сводная информация о выбранных геномах представлена в Таблице 1.
| Таблица 1. Отобранные представители бактерий | ||
|---|---|---|
| Название | Мнемоника | Штамм |
| Bacillus anthracis | BACAN | A0248 |
| Clostridium tetani | CLOTE | 12124569 |
| Enterococcus faecalis | ENTFA | 62 |
| Finegoldia magna | FINM2 | ATCC 29328 |
| Geobacillus kaustophilus | GEOKA | HTA426 |
| Lactobacillus acidophilus | LACAC | La 14 |
| Staphylococcus aureus | STAAR | JH9 |
| Streptococcus pneumoniae serotype 4 | STRPN | AP200 |
В каждом из файлов с расширением .frn было несколько последовательностей 16s-рРНК (копии гена). Выбранные последовательности были собраны в единый файл в fasta-формате (файл можно скачать по ссылке). Названием каждой последовательности является мнемоника соответствующей бактерии.
Выравнивание было построено при помощи сервера MUSCLE. Файл с выровненными последовательностями доступен по ссылке.
Файл с выравниванием был импортирован в программу MEGA методом «Analyze», после чего по нему было реконструировано дерево методом Maximum Likelihood. Результат работы программы представлен на Рис. 1.

Рис 1. Изображение дерева, реконструированного программой MEGA методом Maximum Likelihood.
Реконструированное дерево отличается от правильного. Из всех нетривиальных ветвей только ветви , и <(STRPN, ENTFA), LACAC>были реконструированы правильно. Соответственно, поскольку всего у данного дерева 5 нетривиальных ветвей, то 2 ветви были реконструированы неверно.
Построение и анализ дерева, содержащего паралоги
В этом задании надо было найти в своих бактериях достоверные гомологи белка CLPX_BACSU. были использованы файлы, лежащие в директории P:\y15\term4\Proteomes (они содержат скачанные из Uniprot полные протеомы выбранных бактерий).
Командой cat file1 >> file2 файлы были соединены вместе в один файл. Затем был проведен поиск гомологов программой blastp с входной последовательностью данного белка по базе данных — полученному файлу с протеомами. При поиске был взят порог E-value 0,001. При поиске были использованы следующие команды:
- makeblastdb -in bacs4db.fasta -dbtype prot -out db.fasta
- blastp -query CLPX_BACSU.fasta -evalue 0.001 -db db.fasta -outfmt 6 -out blastres.fasta
В выдаче blastp всего 45 находок, из них 6 принадлежат белку CLPX различных бактерий. В выдаче также 10 находок — по 2 находки, принадлежащие белку HSLU, на каждую из бактерий BACAN, ENTFA, GEOKA, LACAC, STAAR. Остальные находки представлены в единственном экземпляре.
Для дальнейшей работы нужно было из всех последовательностей в файле с протеомами выбрать только нужные 45. Для этого был написан скрипт, отбирающий последовательности по их ID в файле с находками blast’a и записывающий их в новый файл. Полученный файл был импортирован в программу MEGA, затем его последовательности были выровнены при помощи Muscle. По выравниванию методом Maximum Likelihood было построено филогенетическое дерево (Рис. 2).
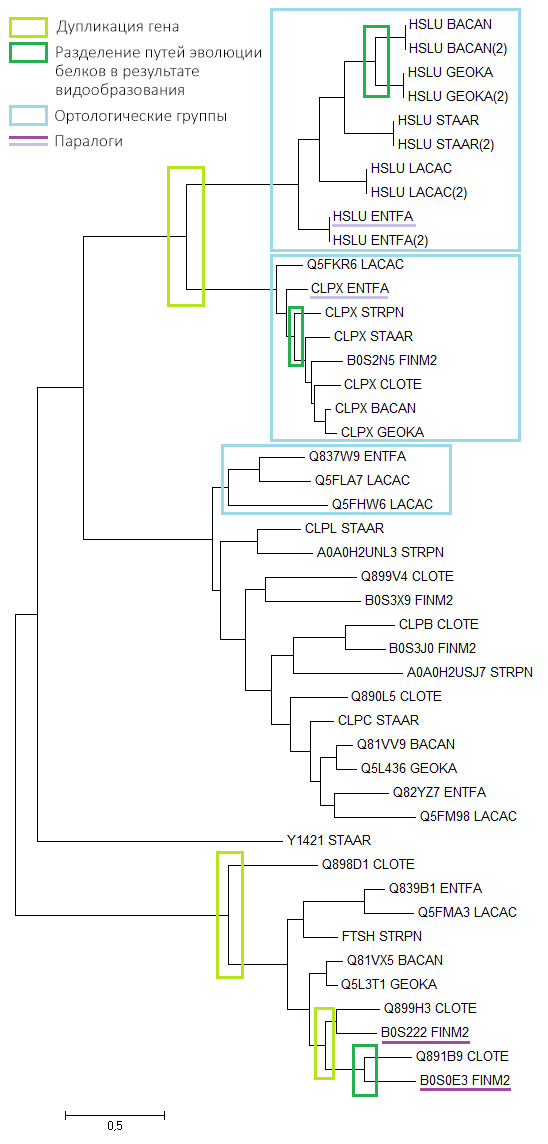
Рис 2. Филогенетическое дерево последовательностей гомологов белка CLPX_BACSU, реконструированное методом Maximum Likelihood в программе MEGA.
На полученном дереве были выделены следующие эволюционные события (при предположении, что дерево было реконструировано верно):
- Дупликация гена: два разных белка выполняют разные функции, но присутствуют у всех/почти всех видов. Например, белки CLPX и HSLU представляют собой разные субъединицы АТФ-зависимой протеазы и оба встречаются почти у всех видов.
- Разделение путей эволюции белков в результате видообразования: такое разделение можно считать выделением групп ортологов.
Также на дереве были указаны:
- Ортологические группы: два гомологичных белка будем называть ортологами, если они: а) из разных организмов; б) разделение их общего предка на линии, ведущие к ним, произошло в результате видообразования. Рамками выделены группы, внутри которых белки попарно являются ортологами.
- Паралоги: два гомологичных белка из одного организма.
© 2014 — 2019 Аксенова М.А. Факультет биоинженерии и биоинформатики МГУ
Источник
Сайт Волынкиной Инны
Для построения филогенетического дерева я выбрала 8 следующих бактерих. В таблице 1 справа представлены обозначения видов, которые я буду использовать в дальнейшем.
| Таблица 1. Выбранные бактерии | |
| Название | Мнемоника |
| Clostridium botulinum | CLOBA |
| Clostridium tetani | CLOTE |
| Finegoldia magna | FINM2 |
| Lactococcus lactis subsp. cremoris | LACLM |
| Listeria monocytogenes serovar 1/2a | LISMO |
| Staphylococcus epidermidis | STAES |
| Streptococcus pyogenes serotype M1 | STRP1 |
| Streptococcus pneumoniae serotype 4 | STRPN |
Скобочная формула дерева
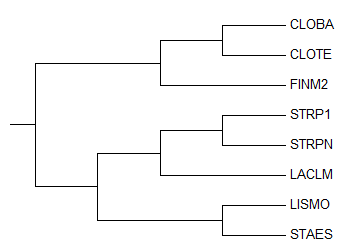
Руководствуясь более полным филогенетическим деревом, я составила следующую скобочную формулу для выбранных организмов.
(((CLOBA, CLOTE), FINM2), (((STRP1, STRPN), LACLM), (LISMO, STAES)));
Изображение дерева
С помощью программы MEGA я построила изображение филогенетического дерева для выбранных мною видов бактерий.
Нетривиальные ветви дерева
Вспомним, что веть называется нетривиальной, если она разбивает множество всех листьев на два подмножества, в каждом из которых более одного элемента — листа.
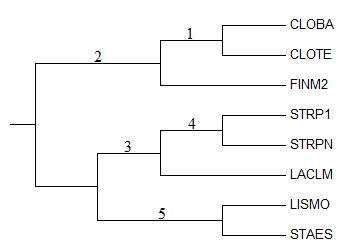
Тогда для полученного мною дерева можно записать слудующий набор нетривиальных разбиений:
- (CLOBA, CLOTE) vs (FINM2, STRP1, STRPN, LACLM, LISMO, STAES)
- (CLOBA, CLOTE, FINM2) vs (STRP1, STRPN, LACLM, LISMO, STAES)
- (CLOBA, CLOTE, FINM2, LISMO, STAES) vs (STRP1, STRPN, LACLM)
- (CLOBA, CLOTE, FINM2, LACLM, LISMO, STAES) vs (STRP1, STRPN)
- (CLOBA, CLOTE, FINM2, STRP1, STRPN, LACLM) vs (LISMO, STAES)
В результате имеем всего пять нетривиальных ветвей:
Полученная картина согласуется с формулами для подсчета количества ветвей дерева, представленными в таблице 2.
| Таблица 2. Число ветвей дерева | ||
| Число тривиальных ветвей (число листьев) | Всего ветвей | Число нетривиальных ветвей |
| n | 2n — 3 | n — 3 |
Реконструкция филогении
Воспользовавшись таксономическим сервисом NCBI, я составила таблицу 3 с перечнем таксонов выбранных бактерий.
| Таблица 3. Таксономия выбранных бактерий | ||||||
| Мнемоника | Тип | Класс | Порядок | Семейство | Род | Вид |
| CLOBA | Firmicutes | Clostridia | Clostridiales | Clostridiaceae | Clostridium | Clostridium botulinum |
| CLOTE | Firmicutes | Clostridia | Clostridiales | Clostridiaceae | Clostridium | Clostridium tetani |
| FINM2 | Firmicutes | Tissierellia | Tissierellales | Peptoniphilaceae | Finegoldia | Finegoldia magna |
| LACLM | Firmicutes | Bacilli | Lactobacillales | Streptococcaceae | Lactococcus | Lactococcus lactis подвид Lactococcus lactis subsp. cremoris |
| LISMO | Firmicutes | Bacilli | Bacillales | Listeriaceae | Listeria | Listeria monocytogenes подвид* Listeria monocytogenes EGD-e |
| STAES | Firmicutes | Bacilli | Bacillales | Staphylococcaceae | Staphylococcus | Staphylococcus epidermidis |
| STRP1 | Firmicutes | Bacilli | Lactobacillales | Streptococcaceae | Streptococcus | Streptococcus pyogenes подвид Streptococcus pyogenes serotype M1 |
| STRPN | Firmicutes | Bacilli | Lactobacillales | Streptococcaceae | Streptococcus | Streptococcus pneumoniae подвид** Streptococcus pneumoniae TIGR4 |
* Listeria monocytogenes EGD-e и Listeria monocytogenes serovar 1/2a — один и тот же подвид. Просто в разных источниках его по-разному обозначают.
** Streptococcus pneumoniae serotype 4 и Streptococcus pneumoniae TIGR4 — один и тот же подвид.
Основываясь на данных из приведенной выше таблицы 3, я подписала нетривиальные ветви дерева, выделяющие определенные таксоны.
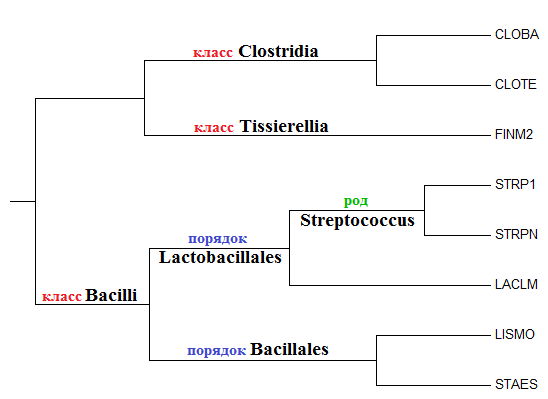
Заметим, что все выбранные бактерии относятся к одному типу Firmicutes .
Источник
Phylogenetic Tree Construction Made Easy with Blast & Mega

Your DNA sequence can be put to good use fairly easily with Blast and Mega software. These programs can help in phylogenetic tree construction. You can ask questions like what is the evolutionary relationship between a set of sequences from different species? Or how have certain microbial strains arisen?
Blast
As any bioscientist probably knows, your first step with a new sequence would be to use BLAST, the Basic Local Alignment Search Tool. This nifty yet powerful resource matches your sequence to the millions of sequences stored in genomic and nucleotide databases. The tool comes up with the sequences most similar to yours. It also gives insights as to the possible identity of those sequences. The results include homologues across species and in similar tissues. Blast is important as it helps to confirm that sequences are homologues and not just lucky alignments.
The basics of using BLAST for nucleotide sequence searches has already been covered in this wonderful article. Below is a brief introduction to the relevant flavors of BLAST found on the NCBI site:
- BLASTN: Compares your nucleotide sequence to the nucleotide sequences in GenBank, NCBI’s repository for nucleotide sequences.
- BLASTX: Compares the six different translation frames (open reading frames) of your nucleotide sequence to the amino acid sequences in NCBI’s Protein Database. This is a great way to find out the possible products and functions of your sequence!
- MegaBLAST: Compares your sequence against other nucleotide sequences, optimal for finding very similar sequences of putatively related species. It casts a tighter net.
Multiple Sequence Alignment (MSA)
Multiple homologues detected via Blast can be aligned using algorithms such as ClustalW or MUSCLE. I like to use MEGA (Molecular Evolutionary Genetics Analysis) because it contains these and other functions. As such it is a one stop source for phylogenetic tree construction.
To start aligning your sequences, launch the Alignment Explorer by selecting the Align | Edit/Build Alignment. This is located on the launch bar of the main MEGA window. From the Alignment Explorer main menu, go to Web-> Query GenBank. This lets you add one by one the sequences for your alignment into the visual explorer. After adding all the sequences, you the option to align them using one to two different programs that are commonly used. You can use ClustalW or MUSCLE software.
Which One to Choose, ClustalW or MUSCLE?
The two alignment programs differ in their operation. ClustalW uses a progressive algorithm for alignment. It aligns two sequences at each step, then aligns the alignment with another sequence, and so on. MUSCLE stands for MUltiple Sequence Comparison by Log-Expectation. It achieves better results than ClustalW across key parameters. These parameters include alignment accuracy as well as lower time and space complexity using progressive, rather than an iterative, alignment.
Go to Alignment, and choose Align by Muscle. As a beginning user, the presets are fine to use, as they serve the purpose of most people. Your output should look something like that shown below.

Save your alignment as a .meg file. This way, you can use it later without having to spend time adding and aligning sequences again.
Phylogenetic Tree Construction with MEGA Version 6
Now comes the fun part! MEGA has a variety of options for phylogenetic tree construction, including UPGMA tree, Maximum Parsimony, Neighbor-Joining, and Maximum Likelihood. These are various approaches to tree construction, each with their own pros and cons, and suitability for your particular purpose. For a given method chosen, Mega will help you find the best model for your DNA or protein sequence substitution rates.
To construct a phylogenetic tree, close the alignment explorer and go back to the Main MEGA Window. We’re going to be constructing a Neighbor-Joining Tree for a quick look at our sequences and their relation to each other. You can always go back and redraw the tree using other methods!
Choose Phylogeny- Construct/Test Neighbor-Joining Tree, and choose your saved .meg file from the Alignment Explorer in the opened dialog box. After choosing and clicking Compute, you get a Tree that looks something like this:

To make that a little easier to read (shown below), click on the button above (Display Only Topology).

Tree Explorer
This tree gives us a lot of information about the sequence! You say, “like what?” It’s now evident that the Zaire Ebolavirus sequence from Gueckedou in Guinea, is most similar to the Mayinga strain (sequence AF272001.1). Both of these are most similar to the strain from Gabon, similar to the ones from Tai Forest or Sudan. This is a surprising fact, considering their geographical locations in Africa. Guinea is in north Africa and Gabon lies across the Gulf of Guinea. This suggest that bats may be an important transmitter of the ebolavirus between these locations.
BLAST and MEGA will help you get a start on analyzing your genome and making sense of the sequence data. This has been a very brief introduction to the power of MEGA. Note, the reliability of the tree can be estimated using the bootstrap method. Happy exploring!
References:
1. Stecher, G., Liu, L., Sanderford, M., Peterson, D., Tamura, K., & Kumar, S. MEGA-MD: molecular evolutionary genetics analysis software with mutational diagnosis of amino acid variation. Bioinformatics 30, no. 9 (2014): doi:10.1093/bioinformatics/btu018.
Источник